Chronic progressive external ophthalmoplegia
Autores: Roberto Rodríguez Rivas y Adib Jorge de Saráchaga
Instituto Nacional de Neurología y Neurocirugíaa¿Por qué leer?
El Dr. Hussain describe un caso clínico compatible con oftalmoplejía externa progresiva y hace un análisis de los primeros casos descritos en 1900, con los cuales orienta a una entidad clínica distinta de las lesiones de sistema nervioso central conocidas como causas de oftalmoplejía en la época.
¿Qué hicieron?
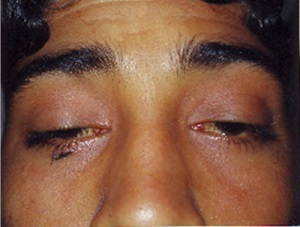
Se detalla a un masculino de 39 años, originario de Rumania, sin antecedentes médicos, que inicia con ptosis palpebral a los 36 años, de forma asimétrica y predominio izquierdo. Se descarta el diagnóstico de miastenia gravis con prueba de neostigmina + atropina, y se observa la limitación de ángulos hacia distintas direcciones oculares, siendo predominante la alteración de ambos rectos externos, de manera asimétrica. Cabe destacar que el paciente no se quejaba de visión doble.
¿Qué aporta?
Los primeros casos descritos fueron por Fuchs (1890) y Wilbrand y Sanger (1900), sin embargo, postularon un daño predominante en el núcleo del tercer nervio craneal y el núcleo de Edinger-Westphal. La degeneración observada por histopatología no era notoria. Más adelante, se observaron otros casos similares, aunque algunos con patología cardiovascular, vistos por Atland (1909), Langdon y Cadwalader (1928) y Farias (1941). Hasta 1946, el profesor Sandifer realiza una biopsia del músculo recto externo en un paciente con oftalmoplejía, ptosis y alteraciones cardiovasculares, notando cambios de degeneración miopática, además de observar una herencia posiblemente materna. Poco a poco, se empezó a retirar a este diagnóstico como una entidad de sistema nervioso central y abordar como una miopatía mitocondrial, diagnóstico en la actualidad que cuenta con más de 20 mutaciones en DNA mitocondrial y/o nuclear.